

Written by Ezzah, Pharmaceutical Research Scholar, focused on practical, reproducible genomics workflows.
Introduction
The genome is not a neat instruction manual. It’s closer to a massive codebase with decades of legacy quirks, undocumented side effects, and a comments section written by evolution at 3 a.m.
So when a model shows up that can look at one million DNA letters at once and predict regulatory signals at single–base-pair resolution, you pay attention. Not because it “solves biology” (it doesn’t), but because it changes what’s practical on a normal research timeline. AlphaGenome was built for exactly that: predicting how sequence relates to regulation, across many modalities, without forcing you to choose between long context and fine detail.
Table of Contents
1. AlphaGenome Open Source: What Changed Overnight (And Why It Matters)
Here’s the fast version: AlphaGenome Open Source is real in the way engineers mean it, the code is public, the weights are available under gated terms, and there’s a ready-to-use API path for people who don’t want to become part-time CUDA archaeologists.
That mix matters. It means you can read the implementation, reproduce key behaviors, and actually stress-test claims instead of arguing about screenshots and hype threads.
1.1 What’s Actually “Open” Right Now
AlphaGenome Open Source Components
| Component | What You Get | What It Enables | What To Watch |
|---|---|---|---|
| Research code | Public repo, installable package | Inspect, run, modify, reproduce pipelines | You still need the right GPU stack |
| Model weights | Downloadable from hosted sources after accepting terms | Real inference, real predictions | Terms are not “do anything forever” |
| API layer | A supported way to call the model | Quick experiments, fewer hardware headaches | You trade control for convenience |
| Datasets and evaluation artifacts | Publicly described, with loaders and metadata | More reproducible benchmarking | Some sources have licensing constraints |
If you’re new to this space, this table is the core mental model: open code is not the same as open weights, and “public” is not the same as “commercially unrestricted.” AlphaGenome Open Source lives in that modern middle ground.
2. What AlphaGenome Actually Does (In One Diagram Worth Of Words)
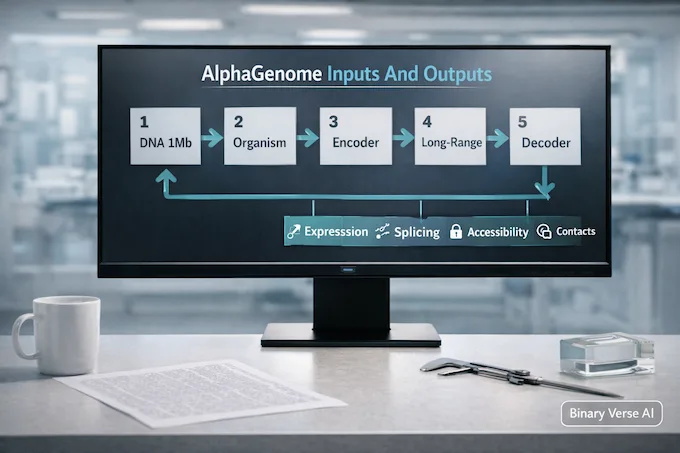
Think of AlphaGenome as a sequence-to-function model that takes a long DNA window and outputs predicted functional genomics signals. Concretely:
- Input: up to 1 Mb DNA sequence (one-hot encoded) plus an organism index (human or mouse).
- Output: predictions across many modalities, including gene expression, transcription initiation, chromatin accessibility, histone marks, transcription factor binding, chromatin contact maps, and detailed splicing signals.
It’s not “a genome model” in the poetic sense. It’s a big, disciplined predictor: if you give it sequence, it gives you predicted tracks. If you give it a variant, it compares REF vs ALT predictions and gives you a structured guess about molecular impact.
A detail worth holding onto: the model predicts thousands of tracks across cell types and assays, including 5,930 human tracks and 1,128 mouse tracks across 11 modalities. That’s not window dressing, that’s why it’s useful when you want to ask, “Is this variant messing with splicing, expression, accessibility, or all three?”
And just to keep your feet on the ground, AlphaGenome Open Source is not a clinical diagnosis machine. It predicts molecular signals, not medical outcomes.
3. Why This Is Significant (The Real Leap Vs The Hype)
Most previous models made you pick two of three:
- Long sequence context
- High resolution
- Many modalities
AlphaGenome pushes hard on all three. The big headline is the 1 Mb context window with outputs that can reach single base-pair resolution for key tracks.
Why does that matter? Because biology plays dirty. A variant can sit far from a gene and still matter through regulatory wiring, chromatin structure, and enhancer–promoter interactions. Long context lets the model “see” more of that wiring in one shot.
And yes, the non-coding story is the actual story. Only a small fraction of human DNA codes for proteins. The rest is regulation, timing, and tissue specificity. AlphaGenome was built to model that regulatory layer at scale, which is why AlphaGenome Open Source has people whispering “AlphaFold moment.” The vibe is similar. The validation reality is not. This is predictive modeling, not a solved physical system.
If you want one practical takeaway: ai for genomics is finally moving from “cool demos” to “tools you can run and argue with.”
4. Open Source, But With Terms: What You Can Use Freely (And What’s Restricted)
Let’s be blunt because it saves time: AlphaGenome Open Source does not mean “free for any business use.” The typical split looks like this:
- Code can be open under a permissive license.
- Model parameters can be gated under non-commercial terms.
- Derivatives often inherit the same restrictions.
That’s not unique to AlphaGenome. It’s the current compromise between scientific openness and model governance. If you’re doing academic research, nonprofit work, or journalism, you’re usually in the safe zone. If you’re trying to plug this into a product pipeline, you need to read the terms like you mean it.
Reproducibility still improves massively, even with gated weights, because the important pieces are now testable: installation, inference behavior, scoring logic, and evaluation scripts.
5. Local Vs API: Choose The Right Path (Most People Should Start Here)
Before you think about GPUs, ask a simpler question: what’s your goal?
5.1 The Practical Decision Rule
- Use the alphagenome api if you want quick iteration, don’t have a data-center GPU, or just want results today.
- Run locally if you need privacy, control, repeatability under your own environment, or you’re working with sensitive sequences.
If your biggest question is how to run alphagenome locally, your second biggest question should be: do you actually need to? Because setup time is real, and your future self will remember every driver mismatch.
That said, if your data must stay in-house, AlphaGenome Open Source plus local inference is the cleanest “data stays with you” story you can tell.
6. Hardware Requirements: Reality Check (And What Works Without An H100)

The official guidance is straightforward: inference is recommended on an NVIDIA H100 class GPU. This is the famous alphagenome h100 requirement you’ll see repeated for a reason. Long context plus lots of heads plus big outputs equals real memory pressure.
Also, the published setup reflects serious compute: pretraining used TPU pods, and distillation was done on H100 GPUs.
That doesn’t mean you can’t run it elsewhere. It means you should expect trade-offs.
6.1 What Breaks First On Smaller GPUs
- Out-of-memory (OOM) when the window is large and outputs are heavy.
- Long XLA compile times the first time you run a path.
- Slow throughput that turns experiments into overnight jobs.
6.2 Practical Hardware Tiers For AlphaGenome Open Source
AlphaGenome Open Source Hardware Setups
| Setup | What You Can Expect | Best Use Case |
|---|---|---|
| H100 80GB | Smooth inference, reasonable speed | High-throughput variant scoring |
| 40–48GB GPU (A6000, A100 40GB) | Might run with careful settings, expect compromises | Focused regions, smaller batches |
| 24GB GPU | Risk of OOM, lots of tuning | Debugging, sanity checks, tiny runs |
| No big GPU | API path is your friend | Exploration, learning, early prototyping |
This is the honest version of alphagenome hardware requirements: you can experiment on less, but you’ll pay in patience.
7. Local Install Step-By-Step (Clean, Reproducible Setup)
This section is your alphagenome installation checklist. The goal is boring success, not heroic troubleshooting.
7.1 Create A Clean Environment
AlphaGenome Open Source environment setup commands
python3 -m venv .venv
source .venv/bin/activate
python -m pip install --upgrade pip7.2 Clone And Install The Repo
AlphaGenome Open Source clone and install commands
git clone https://github.com/google-deepmind/alphagenome_research.git
pip install -e ./alphagenome_research7.3 JAX With CUDA, The Part That Eats Weekends
This is the make-or-break moment. If you remember one phrase, make it alphagenome jax install.
First, confirm your NVIDIA driver and CUDA version. Then install JAX that matches your CUDA stack. One common pattern (CUDA 12 example):
AlphaGenome Open Source JAX CUDA install command
pip install -U "jax[cuda12]" -f https://storage.googleapis.com/jax-releases/jax_cuda_releases.htmlIf you’re on a different CUDA version, use the matching JAX install line from the official JAX docs. Do not freestyle this part.
7.4 Sanity Checks
AlphaGenome Open Source verification commands
python -c "import jax; print(jax.devices())"
python -c "import alphagenome_research; print('ok')"If jax.devices() shows a GPU, you’re past the hardest wall. If it shows CPU only, stop and fix JAX before you do anything else.
This is where AlphaGenome Open Source feels either delightfully normal or deeply cursed. The difference is almost always CUDA and JAX alignment.
8. Download Model Weights (Kaggle Or Hugging Face) And Verify You’re Ready
Weights are available through hosted providers, with terms you accept once, then you download. Keep it simple:
- Accept the model terms.
- Download weights through the supported route.
- Use the provided factory functions.
8.1 Minimal Load Test
AlphaGenome Open Source model loading snippet
from alphagenome_research.model import dna_model
# Pick one:
model = dna_model.create_from_kaggle("all_folds")
# model = dna_model.create_from_huggingface("all_folds")
print("Loaded:", type(model))If that works, AlphaGenome Open Source is actually in your environment, not just in your browser tabs.
9. First Variant Prediction (End-To-End Example You Can Run)
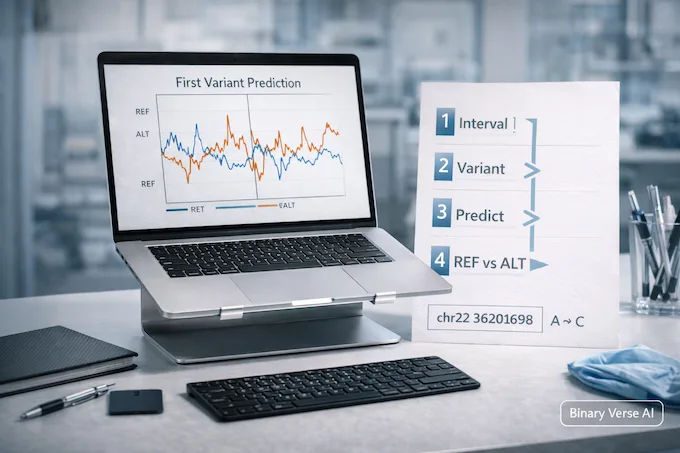
Now the fun part. We’ll run one variant prediction and request a single modality first, RNA-seq, because it’s the easiest output to explain without a PhD in signal processing.
9.1 Minimal Working Example
AlphaGenome Open Source variant prediction example
from alphagenome.data import genome
from alphagenome_research.model import dna_model
model = dna_model.create_from_huggingface("all_folds")
interval = genome.Interval(chromosome="chr22", start=35677410, end=36725986)
variant = genome.Variant(
chromosome="chr22",
position=36201698,
reference_bases="A",
alternate_bases="C",
)
outputs = model.predict_variant(
interval=interval,
variant=variant,
ontology_terms=["UBERON:0001157"],
requested_outputs=[dna_model.OutputType.RNA_SEQ],
)
ref_track = outputs.reference.rna_seq
alt_track = outputs.alternate.rna_seq
print("REF shape:", ref_track.values.shape)
print("ALT shape:", alt_track.values.shape)9.2 Plotting The Difference
AlphaGenome Open Source visualization snippet
import matplotlib.pyplot as plt
from alphagenome.visualization import plot_components
plot_components.plot(
[
plot_components.OverlaidTracks(
tdata={"REF": ref_track, "ALT": alt_track},
),
],
interval=ref_track.interval.resize(2**15),
annotations=[plot_components.VariantAnnotation([variant], alpha=0.8)],
)
plt.show()If you see two overlaid tracks, you’ve done your first real run of AlphaGenome Open Source. That matters more than any benchmark screenshot, because now you can ask your own questions.
10. How To Interpret Results Without Fooling Yourself
This is where most SERPs turn into motivational posters. Let’s not.
A REF vs ALT difference is a prediction about molecular signal, under the assay and tissue context implied by the ontology term and the model’s training distribution. It is not proof. It is not diagnosis. It is not destiny.
10.1 What “Signal Change” Usually Means
- A predicted bump in RNA-seq coverage can suggest altered expression or transcript structure.
- A predicted splicing change can hint at exon skipping or junction shifts.
- A predicted accessibility change can point to altered binding or chromatin state.
All of those are useful because they guide experiments. None of those should be treated as a final answer.
10.2 The Classic Misreads
- Cell or tissue mismatch: you ask the model about the wrong context and blame the model.
- Directionality certainty: the model predicts a change, you assume it’s causal for phenotype.
- Noise blindness: small changes get overinterpreted because plots look convincing.
Use AlphaGenome Open Source like a strong hypothesis engine. Pair it with evidence, QTLs, orthogonal assays, and biological plausibility. That’s how you stay sharp.
11. Troubleshooting: The 8 Errors Everyone Hits
Here’s the short list of pain, and the fix you try first.
- JAX sees CPU only
Fix: reinstall JAX with the right CUDA build, confirm drivers. - CUDA or cuDNN mismatch errors
Fix: align CUDA toolkit, driver, and JAX wheel versions. - XLA compile takes forever
Fix: run once and cache, keep your first test small. - OOM on first real prediction
Fix: reduce requested outputs, shorten intervals, avoid heavy modalities first. - Interval errors, wrong chromosome build
Fix: confirm coordinate system and chromosome naming, chr22 vs 22. - Ontology term issues
Fix: use known-good terms from examples, then expand. - Weights not found or download failures
Fix: confirm terms accepted, ensure credentials work, verify cached files. - Plotting fails on headless servers
Fix: use a non-interactive backend, or save figures to disk.
If you handle these, AlphaGenome Open Source becomes a tool you can trust operationally, not just admire conceptually.
12. Limitations, Ethics, And Privacy (The Part People Argue About)
AlphaGenome is powerful, and still incomplete in predictable ways.
- It does not make biology deterministic.
- It is not validated as a medical device for personal genome prediction.
- It predicts molecular proxies, not full trait causality.
- Very long-range regulation remains hard, and tissue specificity is still a frontier.
Privacy is straightforward in principle: local runs keep control with you, API runs depend on policies you must read. The inequality question is real too. Models like this land first where compute exists, then trickle down. The best counterbalance is open tooling, reproducible workflows, and a community that publishes failures as loudly as wins.
If you’re here because you want ai for genomics to move faster, this is the right kind of work. Predict, test, iterate, share.
12.1 A Simple Next Step
Pick one variant you care about, run it through AlphaGenome Open Source, then do one extra thing that most people skip: write down what evidence would change your mind. If your plan survives that test, you’re doing science, not vibes.
If you found this useful, share it with a labmate, and if you hit a weird install edge case, document it and open an issue or a short write-up. The fastest way to make AlphaGenome Open Source better is to treat it like real infrastructure, because that’s what it’s becoming.
1) What is a regulatory variant?
A regulatory variant is a DNA change that affects how genes are turned on or off, rather than changing the protein sequence itself. These variants often sit in non-coding regions and can shift expression levels, timing, or tissue specificity, which is why they matter so much in ai for genomics.
2) What is the variant effect?
Variant effect is the measurable change a mutation causes in predicted or observed biology, like RNA expression, splicing, chromatin accessibility, or transcription factor binding. In AlphaGenome workflows, it often means comparing predictions for reference vs alternate alleles to see what signal moves, and by how much.
3) What does a regulatory gene mean?
“Regulatory gene” usually refers to a gene whose product controls other genes, like transcription factors or chromatin regulators. People also use the phrase loosely to mean “a gene involved in regulation.” In practice, focus less on the label and more on the mechanism, what it controls, where, and under which cellular conditions.
4) What is a regulatory region mutation?
It’s a mutation in DNA regions that control gene activity, promoters, enhancers, silencers, insulators, and related elements. Instead of altering a protein, these mutations change the “settings” of gene expression. They’re notoriously hard to interpret, which is exactly why long-context models are getting attention.
5) Can I run AlphaGenome locally, and is my data secure if I do?
Yes, you can run AlphaGenome locally if you meet the alphagenome hardware requirements and handle the alphagenome jax install correctly. Local runs keep sequences on your machine, which is the cleanest privacy story. If you use the alphagenome api instead, assume your sequence leaves your environment, then read the service terms and security posture before sending sensitive data.